-

-
-

-
-

-
-

- 010-56548231
-

![]()

![]()

![]()

![]()
![]()
 Sequencher 5—DNA序列分析软件
Sequencher 5—DNA序列分析软件

Sequencher 是 DNA 序列分析的工业标准软件。它可以和所有的自动序列分析仪一同工作,并且因为它的极速 Contig 组装、很短的学习曲线、用户友好的编辑工具,以及卓越的技术支持闻名。
Sequencher 从差不多 15 年前释出第一个版本到现在,Sequencher 在每个基因专业和制药公司中都应用与于序列分析任务,同时在全球超过40个国家里为数众多的学术和政府实验室中使用。Sequencher 被生命科学研究者们使用于许多不同的 DNA 序列分析应用方面,包括基因重组、突变检测、法医的人体辨别,以及分类学等等...
Sequencher 的能力包括杂合子和 SNP 的检测和分析,cDNA 到 染色体 DNA 的大型间隙对准,比较排序,对置信评分的支持、ORF 转换、基因银行特性化导入、以及限制酶映射等等。
SNP 检测 (SNP Detection)
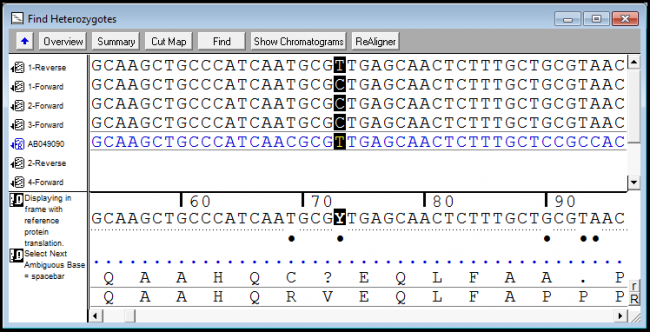
Sequencher 有几个强大的工具可帮助您检测 DNA 序列中的突变和 SNP。您可以使用 Sequencher 在一组序列之间进行比较序列比对,或者将一个或多个序列与参考序列进行比较。Sequencher 的Call Secondary Peaks...功能分析所有序列中的潜在杂合子。很容易控制定义杂合子的严格性。
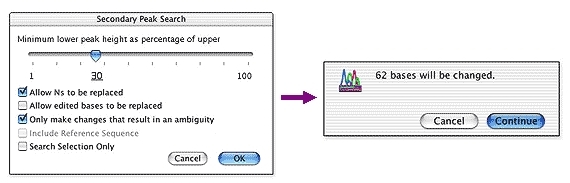
绘制 DNA 组装概览中所有杂合子的位置。
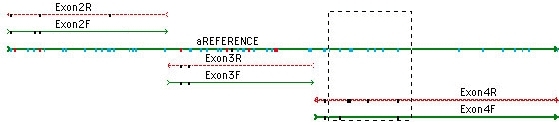
您可以从一个杂合子导航到下一个杂合子;只需单击 Bases 视图中的空格键。查看共有序列和共有序列下方的参考序列的蛋白质翻译。参考序列可确保您的 SNP 编号从一个 DNA 组装到下一个组装是一致的。
序列装配 (Sequence Assembly)
Sequencher 的装配算法可以将你的 DNA 片断快速而又准确的装配起来。自觉的工具允许你在数秒内就可以设置好参数并且调整它们。
你完全可以不顾方向的装配序列。Sequencher 会比较前端和反转补体方向来装配最可能的 Contig。
将 Sequencher 的多功能装配工具应用到:
■确定矢量构造
■装配病毒和细菌基因组
■从 cDNA 库中聚类数以万计的序列
■将基因变体和一个参考序列做对比
■创建一个引物地图
■将 cDNA 装配成基因组序列
以及其他许多项目...

序列编辑 (Sequence Editing)
Sequencher 为您提供 DNA 序列编辑工具,您需要知道序列是绝对正确的。您可以一次查看一个序列的色谱图数据,或以正向和反向查看多个对齐的色谱图。
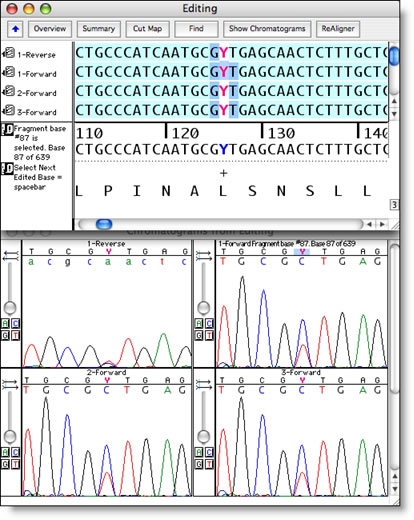
滚动浏览对齐的数据既快速又容易,或者您可以使用 Sequencher 的选择工具突出显示差异或低质量的区域。
自动分析 (Automated Analysis)
Sequencher 可对你的数据进行批处理,这个过程是透明、用户可定义、可恢复的,并且 Sequencher 从不为了自动化的目的而破坏你科学结论的正确性。
当你工作在多个序列时,Sequencher 可以:
■调用第二峰
■调整矢量
■调整低质量的尾端
■创建一致序列
■恢复到实验数据
Sequencher 总是管理两份数据,一份编辑过的,一份则是原始的导入数据。当你应用“Revert to Experimental Data”命令到在你项目中已选定的序列或者在一序列中的选中部分时,你可以撤销所有或者部分编辑动作。
自从 4.5 以后,Sequencher 就包含了一个新的自动化工具,“Assemble by Name”。依靠“Assemble by Name”,你可以选择片段名的一部分作为一个共享的标识符,或者说是“装配句柄”。Sequencher 就可以将选择和名字自动转换为 Contig。例如,仅仅通过一个按钮的点击,你就可以将 90 个文件,45对前端和反向序列转换成 45 个根据你的病人编号命名的 Contig。装配参数的每个变化都会重组你的片段,因此你可以根据克隆编号、日期、引物,或者其他任何你记录在序列名称中的特征,来装配 Contig。
如果你做了许多排序,你会感激“Assemble by Name”,尤其是在你有许多个带有一个标准引物集的样本序列时。事实上,任何在序列名中具有可用信息人的人,都有可能使用“Assemble by Name”。一些应用包括:
■用于身份鉴定和人口研究的 MHC 和线粒体基因排序。
■候选基因的 PCR 产品的分析。
■对保存的跨系统分类物种基因的排序。
■通过跟踪病毒对抗病毒剂或疫苗的抗药性,对病毒序列进行监控。
即使在项目中只有一个 Contig,“Assemble by Name”仍然是有用的,因为它确保 Contig 的名称可以精确的表述样本的名称。如果你有命名为“Bob”的样本,那么正确命名你的 Contig 并不是什么难事,但但你有象“Bob-2309888762-4321”这样更复杂的名字时,允许 Sequencher 命名你的 Contig 是非常有用的。
矢量微调 (Sequence Trimming)
自动化 DNA 测序仪偶尔会产生质量较差的读数,特别是在测序引物位点附近,以及在较长序列运行结束时。DNA 文库中的克隆序列通常包含载体序列、polyA 尾或其他不相关的序列。内含子和引物序列经常位于扩增外显子序列的两侧。除非通过修剪去除,否则任何这些伪影都会扭曲您的序列组装和下游序列分析。
Sequencher 提供了简单易用但功能强大的工具,可帮助您修剪质量差或模棱两可的数据:
Trim Ends从测序片段的末端去除误导性数据。
Trim Vector去除污染序列末端的序列特定数据。
修剪到参考消除了超出组装参考序列的序列末端。
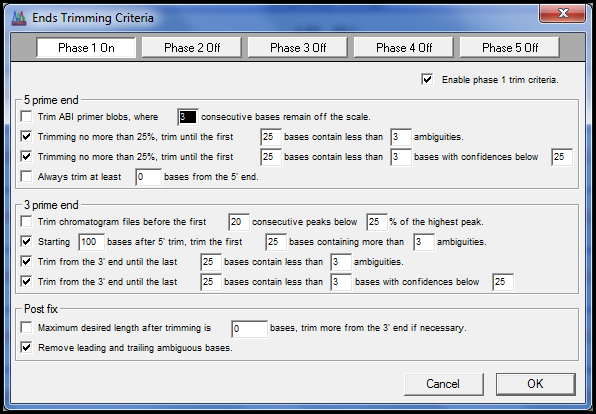
在执行修剪之前,Sequencher 会显示建议修剪的图形表示,这使您可以进一步细化您的标准。
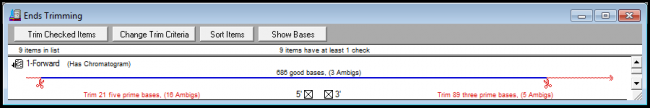
如果您想在修剪序列的任一端或两端恢复一定数量的碱基,因为您的修剪过于严格,或者您想提高覆盖率, Batch Revert Trim Ends 可以让您做到这一点。只需单击几下,您就可以将碱基恢复到只有几个或几千个序列,并更好地控制您的序列修剪。
2016年8月正式发布Sequencher 5.4.5,Sequencher 5.4.5扩大了大型基因组功能。用BWA-MEM和GSNAP插件的BAM文件取代了SAM文件输出。这个小改变有很大影响NGS序列分析的几个方面。首先这大大降低了NGS的存储需求分析。其他NGS工具也将受益于使用BAM文件。BAM文件加载在NGS查看器的程序,如平板电脑会快得多。
以下为英文介绍
News:
Sequencher 5.4.5 is Available Now!
New features for Sanger and Next-Generation DNA Sequence Analysis include:
Sanger:
•New Batch Revert Trim Ends command.
•Ability to adjust the font size in the Project Window.
•For hybrid sequencing projects, you can create NGS reference databases/indexes directly from the consensus sequences in the Project Window in addition to linking to an external sequence.
NGS:
•GSNAP and BWA-MEM workflows now produce BAM files.
•Enhancements to the External Data Browser (EDB).
•FastQC Reports now open automatically.
 北京友万信息科技有限公司,英文全称:Beijing Uone Info&Tech Co.,Ltd ( Uone-Tech )是中国大陆领先的教育和科学软件分销商,已在中国300多所高校建立了可靠的分销渠道。拥有最成功的教学资源和数据管理专家。如需申请软件采购及老版本更新升级请联系我们,咨询热线:010-56548231 ,咨询邮箱:info@uone-tech.cn 感谢您的支持与关注。
北京友万信息科技有限公司,英文全称:Beijing Uone Info&Tech Co.,Ltd ( Uone-Tech )是中国大陆领先的教育和科学软件分销商,已在中国300多所高校建立了可靠的分销渠道。拥有最成功的教学资源和数据管理专家。如需申请软件采购及老版本更新升级请联系我们,咨询热线:010-56548231 ,咨询邮箱:info@uone-tech.cn 感谢您的支持与关注。
地址:北京市昌平区中兴路21号院4号楼5层516 网站备案号:京ICP备16049373号-1]
联系方式:+86-10-56548231
